
CD-HIT是一個應用廣泛的序列比對分群軟體,它可以幫你找到具有代表性的序列,在合理時間內處理巨量的序列資料,大大降低你擁有資料的複雜度,減少後續計算需求。剛推出時CD-HIT著重於針對蛋白質序列的分群,目前對於DNA序列甚至於對處理次世代定序的原始短序列也有相對應的程式可供使用,使它的應用更加廣泛。
舉例來說,假使你使用了序列組裝軟體來處理轉錄體資料,得到了數以萬計的transcripts,仔細檢查後發現這些transcripts中其實彼此間有相當程度的雷同;此情況固然有可能因為生物體中原本即存在不同的transcript isoforms,但也可能是因為原始序列中包含的error callings造成組裝時出現了多種可能路徑而讓最終結果transcripts的種類數量變得龐大;在某些組裝軟體中甚至可能出現同一序列的小片段形成另一條transcript的情況。此時我們可考慮使用CD-HIT來幫助刪除大量極相似的transcripts,使後續的轉錄體分析變得較為合理可行。
使用者可到https://github.com/weizhongli/cdhit/releases下載最新版本的CD-HIT,在LINUX系統下解壓縮後,依照README的簡單指示操作安裝使用。
CD-HIT提供了同一組內序列比較,與兩組間序列比較的功能。當我們對一組蛋白質(或DNA)序列做相似度分群時,可使用套組內的CD-HIT功能(或CD-HIT-EST),並自己設定參數以定義你要的相似程度。同樣的,若想要比對兩組蛋白質(或DNA)序列是否相似時,改為使用CD-HIT-2D(或CD-HIT-EST-2D)。
指令範例:
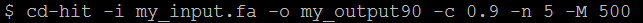
這裡列出幾個比較常用的參數:
-i : 輸入檔案名稱,例如my_input.fa
-o : 輸出名稱,例如my_output90
-c : 相似度設定,例如0.9 = 90%,即相似度高於90%的序列會被當成同一群集。
-n : 框架長度,不同的相似度有其建議的n值設定,例如90%時建議n =5。
-M : 最大記憶體限制,例如500即最高系統記憶體使用量不超過500Mbytes。
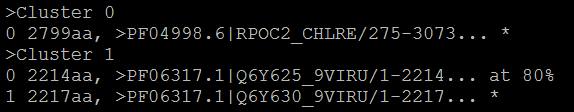
結果會輸出於副檔名為.clstr的文字檔中(例如my_output90.clstr),其內容顯示如下(>後為此群集的名稱,每一條序列都會註明長度,標示在序列後方的*表示此序列為此群集中的代表序列,%為此序列與代表序列的相似程度百分比):
輸出結果同時也包含一個fasta檔案(例如my_output90.fa),檔案中列出所有群集的代表序列(也就是後面附註*的那些),使用者可直接取此檔做後續分析,非常方便。
另外因應次世代定序的發展,新版本的CD-HIT與時俱進,陸續推出CD-HIT-DUP與CD-HIT-454等功能用以處理來自Illumina或454平台的定序結果原始序列,而原本的CD-HIT-EST與CD-HIT-EST-2D功能也可用來處理paired-end的原始序列資料了!
(可參考官方網頁:http://weizhongli-lab.org/cd-hit/)
感謝 TIGP 的 chunyic 同學提供以上資料