
前言:以蛋白質結構為基礎來分析蛋白質與其它蛋白質或小分子結合的特性及位置,進一步了解此一蛋白質的功能。
介紹的資料庫與工具:
1. IBIS (Inferred Biomolecular Interaction Server)
2. PISA (Protein Interfaces, Surfaces, and Assemblies)
IBIS (Inferred Biomolecular Interaction Server)是由NCBI結構組發表的一個探討蛋白質與蛋白質、小分子、核酸之間交互作用的伺服器( Nucleic Acids Res. 2012 40 Database issue: D834-D840) (https://www.ncbi.nlm.nih.gov/Structure/ibis/ibis.cgi)。
這個伺服器包含整理、分析及預測蛋白質其交互作用的夥伴(interaction partners)及結合位點(binding sites),並提供搜尋及比較結構資料庫中同源蛋白質。根據序列和結構的保留性,在同源蛋白質中發現相似的結合位點,經過分析驗證之後進行排序及集群(clustering)。使用者可以快速地得到蛋白質及與其作用的分子及結合位點的交互作用數據及相關訊息。
在IBIS演算過程圖示中,步驟一是以一個已知蛋白質結構來比對資料庫中所有蛋白質結構區域(domain)或結構鏈(chain),並且收集與此詢問蛋白質具有至少30%的序列一致性的相似結構群。步驟二是將結構序列的結合位點進行排列及選取結構上可以排列的蛋白質聚集成群組,並與詢問蛋白質的結合位點對齊。步驟三是為了排除序列中與結合作用無關的位點,應用BLOSUM的評分系統將原始分數轉換為比特得分(bit score),通過除以比特得分的最大值來對相似度得分進行標準化(normalization),使得可以比較來自不同接合面(interface)比對的保守分數(conservation score)。在步驟四中使用完全連鎖集群演算法(complete-linkage clustering algorithm)對同源結構的結合位點進行集群及分類,再利用PISA (Protein Interfaces, Surfaces, and Assemblies)演算法來驗證。步驟五是依據集群的結合位點排列來構建位置特定得分矩陣PSSM (PSSM position specific score matrix),較高的序列PSSM分數意指此一結合位點在生物相關的註解有較高的可能性。最後,將包括詢問蛋白質有關的結合位點PSSM分數、結構比對的序列一致性、接合面的接觸數目(contact number)和結合位點排列的保留列(conserved columns)的分數等等的數據,進行標準化並計算出Z-score,所有的集群(clusters) 再依照Z-score進行排名(ranking score)。如果出現無法定義排名分數的集群,這些集群就稱為“單獨”(singletons)亦即一個集群只有一個成員。
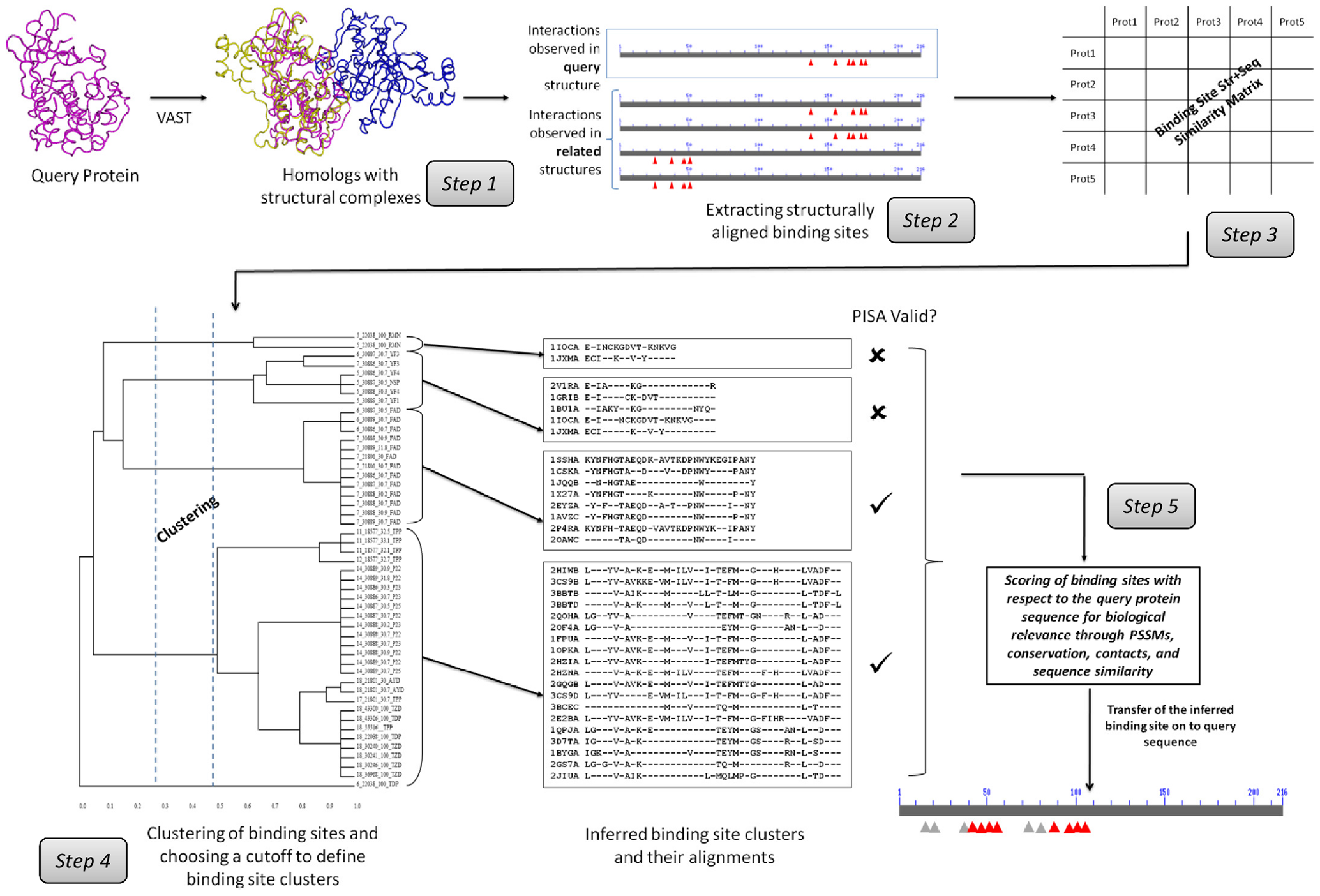
(資料來源:http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028896)
以下的IBIS蛋白質結構範例是PDB代碼5CID,是酵母菌yeast mitochondrial cytochrome C peroxidase (Ccp;鏈A和鏈C)帶有W191G殘基變異和另一蛋白質分子cytochrome C (Cc;鏈B和鏈D)分別結合血紅素heme所形成的複合物。開啟IBIS網頁後在詢問欄中填入5cida,頁面下方則出現搜尋結果,點擊左側網絡圖形或其下方 ”All interactions for query sequence” ,即出現新的視窗來顯示與此一蛋白質有交互作用的其他分子,包括蛋白質、化學分子、核酸和胜肽,甚至也包括離子。點擊圖片中的化學分子或離子會出現PubChem CID代碼及註解,點擊圖片中的蛋白質或區域,則會出現CDD (Conserved Domain Database)資料庫的代碼及註解。接著點擊搜尋結果的任一種分類或IBIS定義的集群數量,來進一步察看[圖一]。
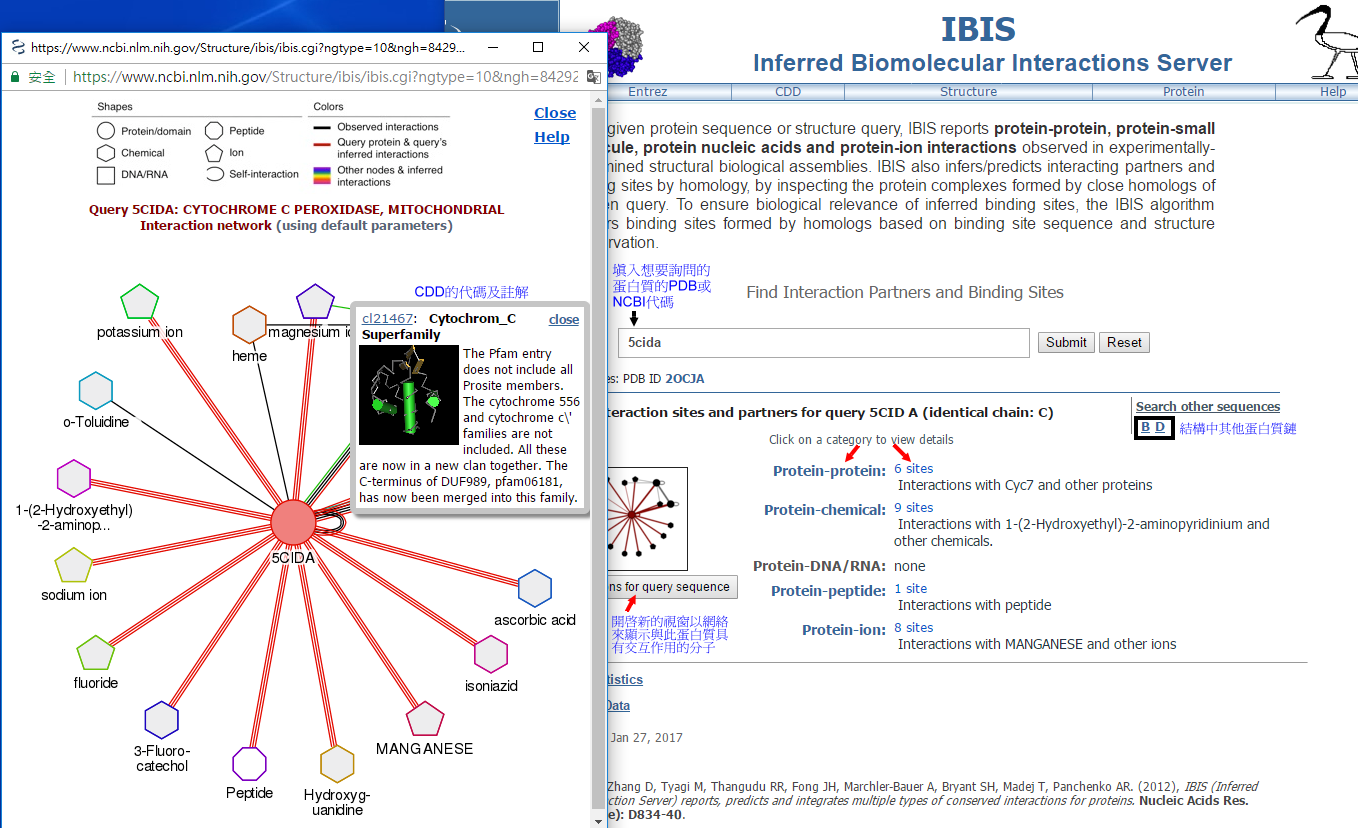
[圖一]
結構範例5cida有6個”protein-protein”結合位點的集群,其中的5個集群是屬於“單獨”(singletons) 集群。右側圖表上方是詢問蛋白質的序列長度及不同顏色的實心橫框來表示CDD定義的區域位置,接著是IBIS註解在各別集群的結合位點,排列於結合位點中的殘基的保留程度以不同顏色的三角形顯示:紅色表示高度保守,藍色表示中等和非保留的殘基以灰色表示,移動滑鼠游標在會三角形上顯示其在Genbank蛋白質序列的殘基數字。
下方則是結合位點的集群以”Ranking Score”由大至小的順序所產生的列表,點擊”+”符號來展開各別集群的資料。資料中第一個集群的結合位點是由20個胺基酸殘基組成,顯現有另外3個同源蛋白質的結構具有此一結合位點,例如PDB代碼4GED鏈A為Leishmania major cytochrome C peroxidase,與5CID鏈A的序列一致性為40%[圖二]。但是這個表格只有列出2個結構,選擇”See all members”聯結至列出所有同源結構的表格,表格中也包含各個結構的蛋白質序列可行與此一集群的結合位點相對應的殘基數量,例如4GED鏈A是18個胺基酸殘基[圖三]。
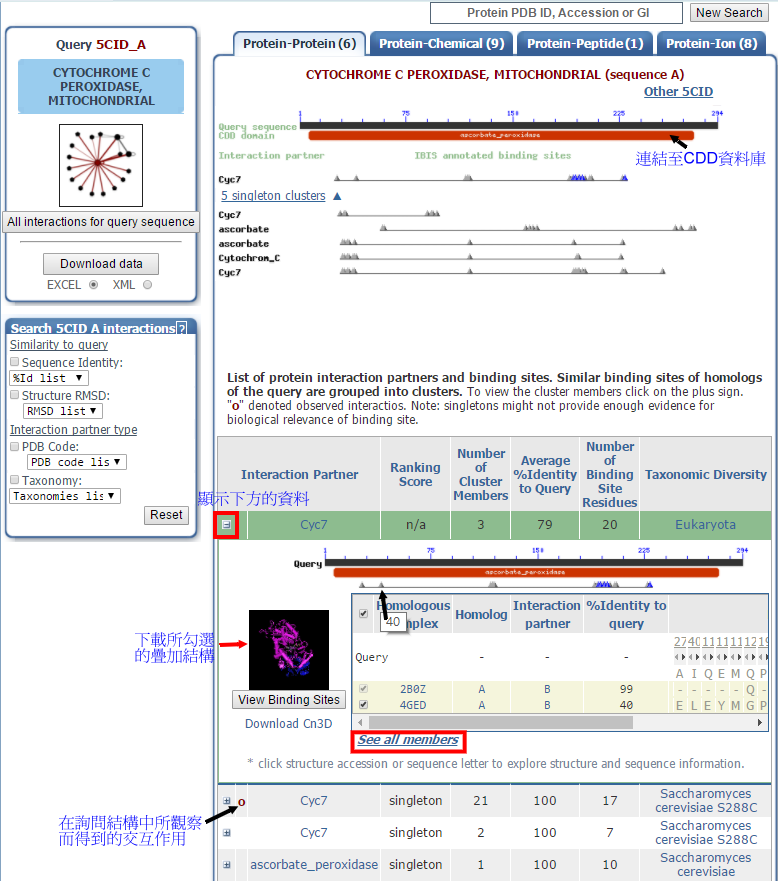
[圖二]
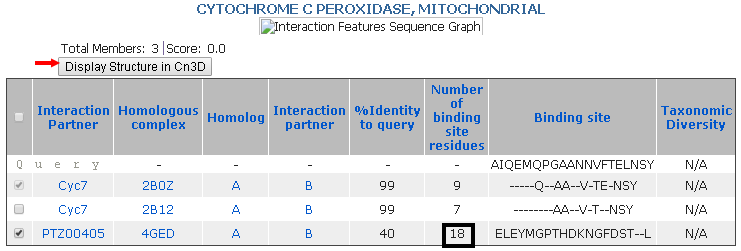
[圖三]
接下來要查看此一集群的結合位點在三維結構中的位置,在圖二中,表格中了第一個同源結構是預設的,會自動被勾選,所以再勾選4GED後,點擊三維結構的圖片或”View Binding Sites”,則三個同源蛋白質的疊加結構 (superimposed structures)將被下載為cn3的檔案格式。圖四中位於5CID鏈A的結合位點的殘基在結視窗顯示為淺紅色sticks形式,在序列排列視窗則顯示為紅色的殘基。使用者可下載Cn3D來檢視,關於Cn3D軟體的詳細操作方法請參照PDB-related databases and tools Part II (https://lsl.sinica.edu.tw/Blog/2017/05/pdb-related-databases-and-tools-at-ncbi-part-ii/)。
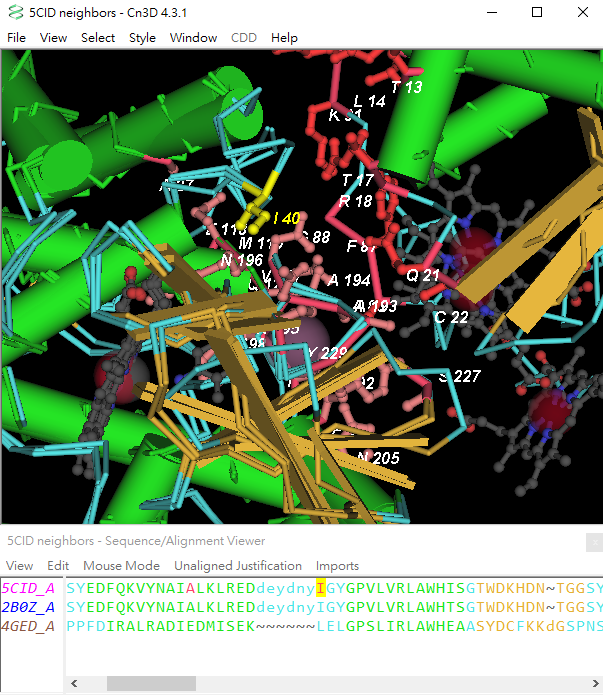
[圖四]
由EBI (European Bioinformatics Institute)所發表的資料庫PISA (J. Mol. Biol. 2007 372:774-797) (http://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver)被認為是識別結構中蛋白質與複合分子之間存在的接合面的最佳方法之一,它是一種自動化的方法並且以晶體結構(crystal structures)報告中的接合面資料和組件(assembling)穩定性的分析來檢測巨分子的組合物(mocromolecular assemblies)。
在PISA的詢問欄中填入結構範例4GED則出現結構的初步資訊,包含2個蛋白質鏈及4個配體(ligands),另外還有4個分子形成最有可能的組合物,亦即在晶體中有兩個鏈A和鏈B結合的複合物[圖五]。點擊Interfaces開啟一個列表,係在此晶體結構中被檢測到的分子間具有交互作用的接合面。
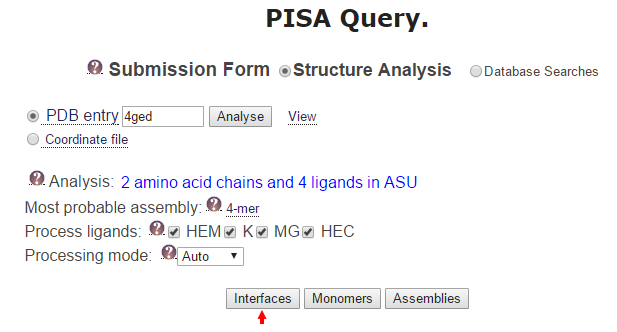
[圖五]
在圖六的列表顯示總共有17個交互作用的接合面被偵測到,其中第4個接合面是由鏈B和鏈A所形成,位於鏈B的結合位點有15個殘基而鏈A有25個殘基參與,這些胺基酸殘基之間形成8個氫鍵(hydrogen bonds)和5個鹽橋(salt bridges)。接著點擊第4個接合面及列表的上方的Details來取得此結合位點的詳細資料。
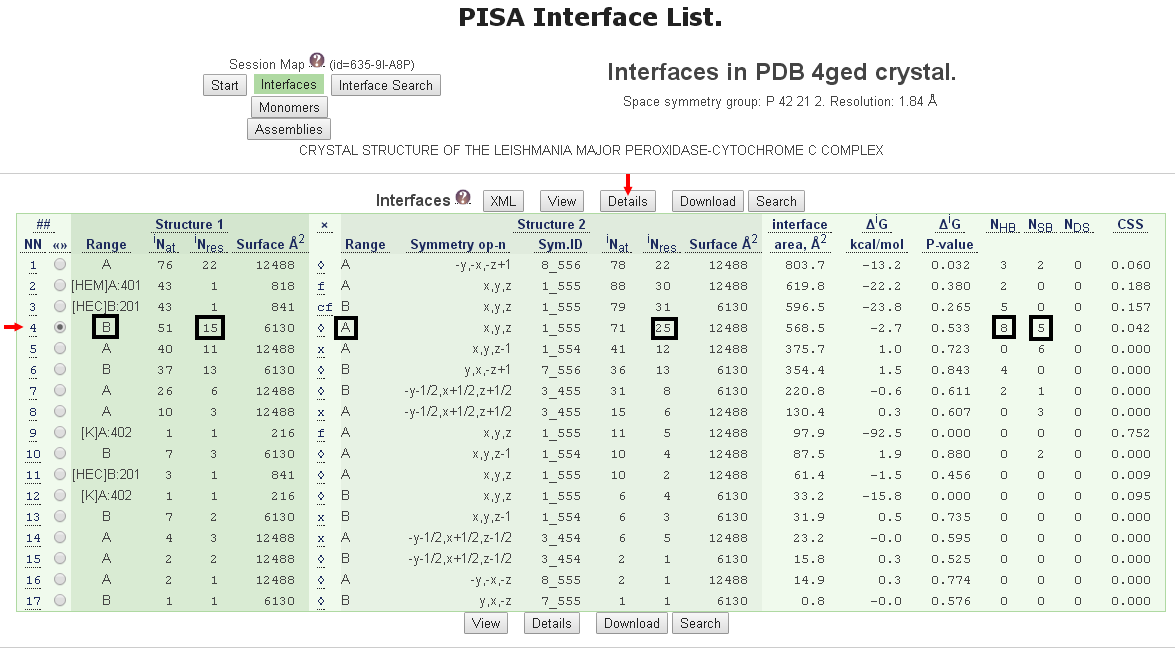
[圖六]
PISA不僅提供接合面殘基非常詳細的資料,也提供包含結構中其他胺基酸的屬性資訊,包括可親近的表面面積(Accessible Surface Area)、隱藏的表面面積(Buried Surface Area)、接合面殘基的溶劑化能量效應(Solvation energy effect)、和隱藏面積的百分比(Buried Area Percentage)等等,另一方面又以HDSC來表示接合面殘基之間氫鍵、鹽橋、雙硫鍵和共價作用的形成與否。在下方的列表也很清楚地以不同顏色的背景來顯示各個殘基在結構中的特性,例如淺藍色是溶劑可親近殘基(Solvent-accessible residues)、藍色是不可親近的殘基(Inaccessible residues)和淺黃色是位於接合面的殘基(Interfacing residues) [圖七]。
PISA的另一項功能是以詢問結構與資料庫中所有複合物結構進行接合面的搜尋,在圖七的上方選擇Interface Search就出現如同圖八的搜尋設定。按壓Submit for search等待數分鐘後,搜尋出23個與鏈B和鏈A接合面相似的複合物結構[圖九],點擊左側的數字,則出現該接合面的資料,例如點擊”1”即回到圖六的資料頁面,這些相似的複合物結構可以提供進一步的分析及研究。
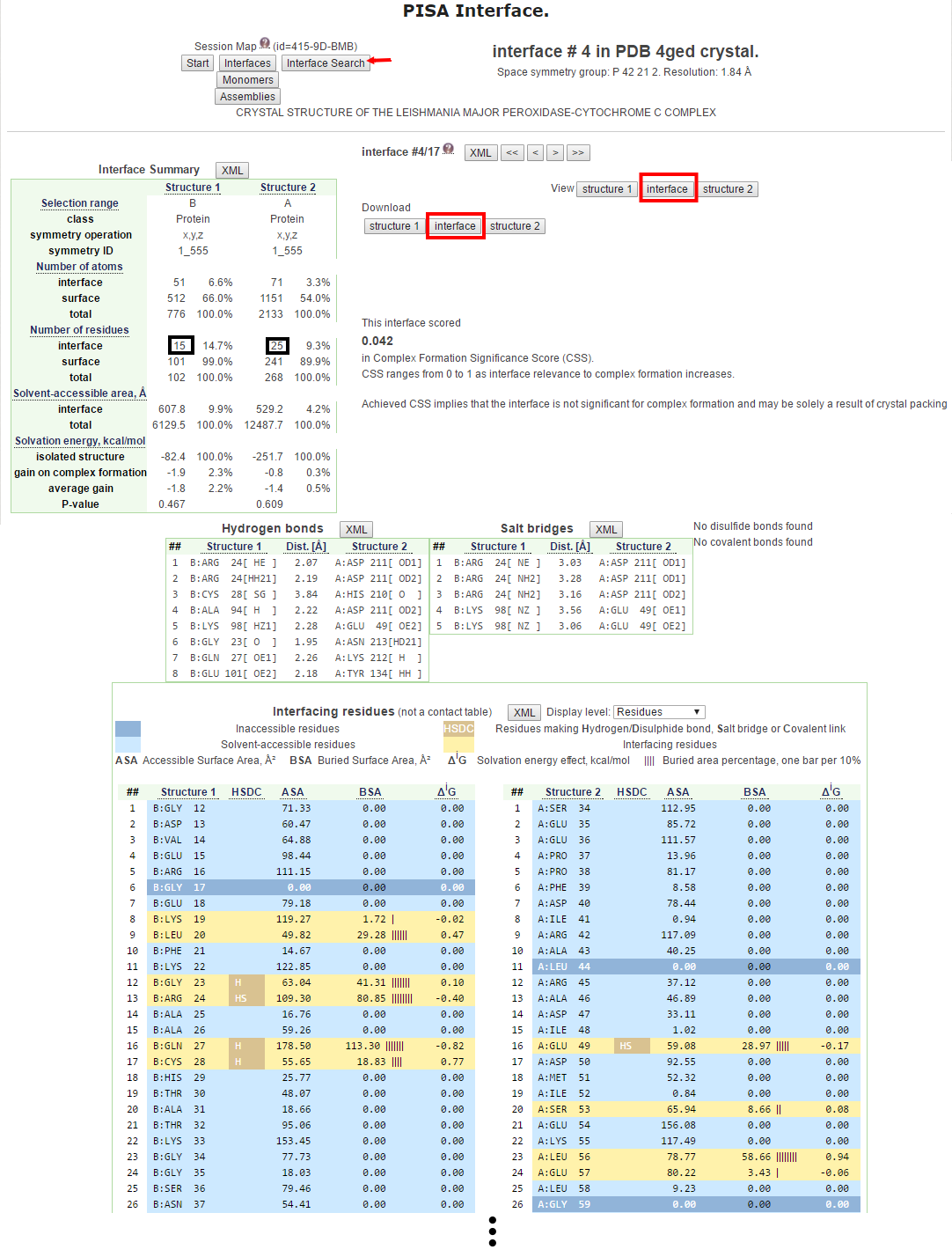
[圖七]
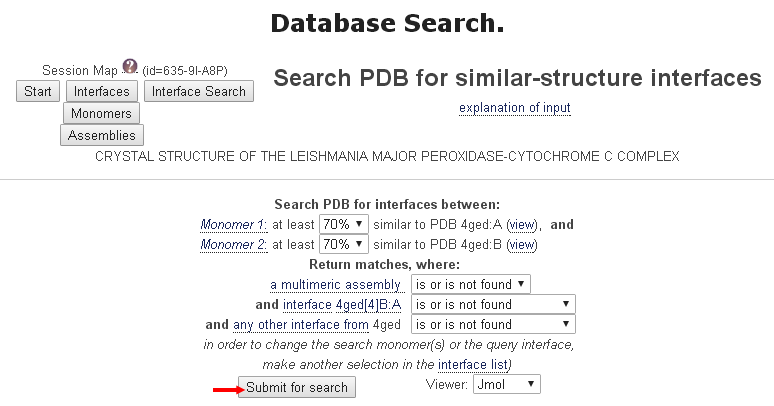
[圖八]
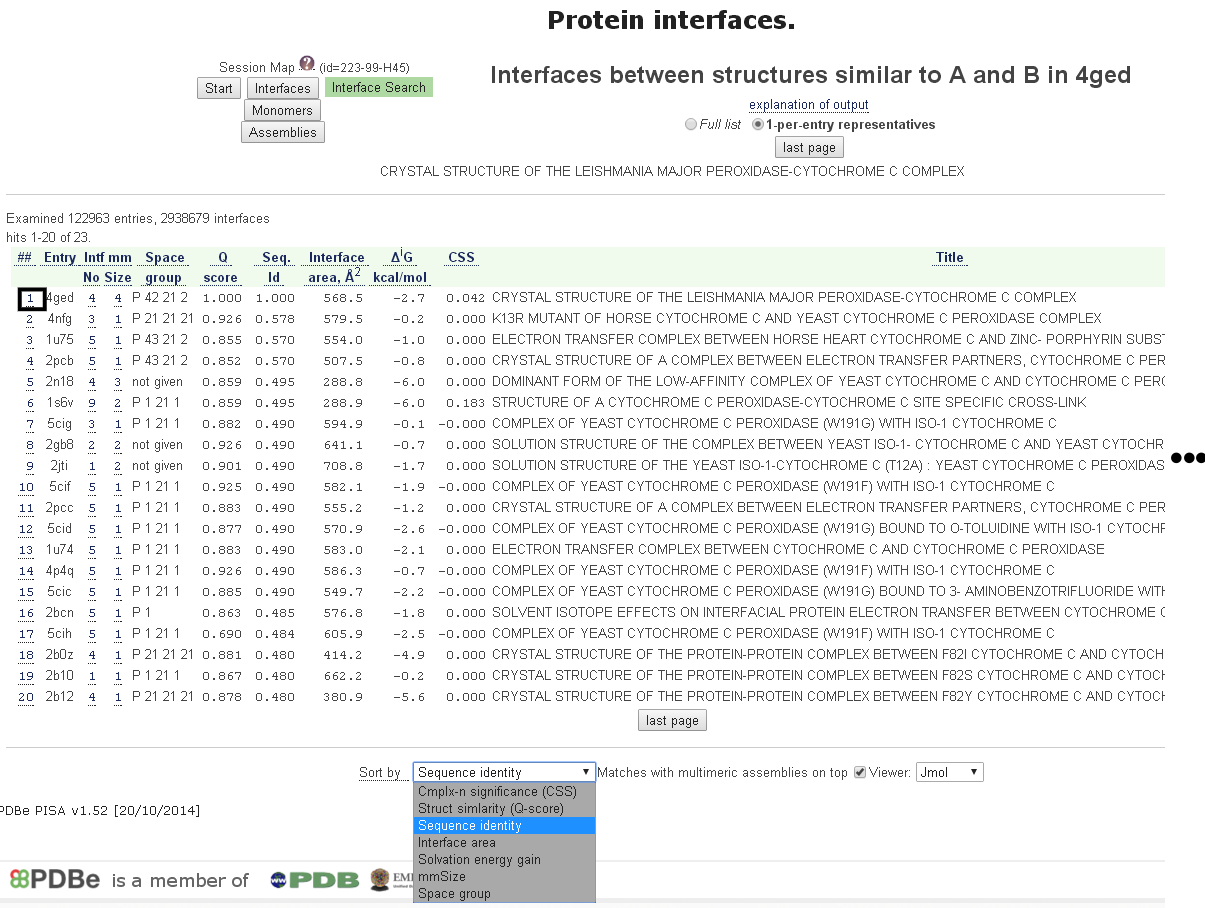
[圖九]
在結構的檢視方面,選擇圖六中的View功能或圖七中View→ interface功能,出現以Jmol設計的結構視窗,藍色的鏈B和青色鏈A顯示為Spacefill的模式,鏈A位於接合面的結合位點為綠色,鏈B則為紅色[圖十]。

[圖十]
或是選擇圖六中的Download功能或圖七中Download→interface功能,下載兩個蛋白質鏈的結構再運用其他結構檢視工具例如Rasmol或Swiss-PdbViewer。圖十一是使用Rasmol版本2.7.5.2標示出鏈B和鏈A接合面的接合殘基,並分別計算位於鏈A Glu49 (OE2)和鏈B Lys98 (NZ)原子的距離(distance)為3.06Å及鏈A Asp211 (OD1)和鏈B Arg24 (NE)為3.03Å[圖十一]。Rasmol的操作如下:
1. 編輯 script檔案 (script01.spt)

2. 編輯 script檔案 (script02.spt)

3. 開啟Rasmol後,選擇Open打開script01.spt檔案→選擇Display項目的Ball & Stick→選擇Open打開script02.spt檔案→選擇Settings項目的Pick Monitor→在結構上點擊原子即可。
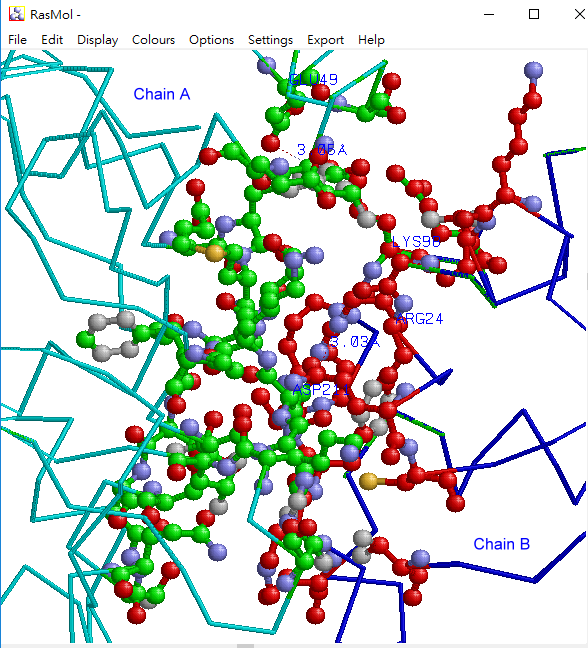
[圖十一]
PISA不但提供詳細的分子間交互作用的資料,也提供巨分子的生物單元及組合物的資料,例如病毒顆粒(virus particle)的組成結構。圖十二和十三是以病毒Coxsackievirus A16(PDB代碼5C4W)為例子,基本結構組成是鏈A(vp1,黃色)、鏈B(vp2,綠色)、鏈C(vp3,青色)和鏈D(vp4,紅色)[圖十三左側],下載5C4W的組合物(20和240單元)並以Rasmol檢視其結構的五聚體和顆粒結構[圖十三中間及右側]。
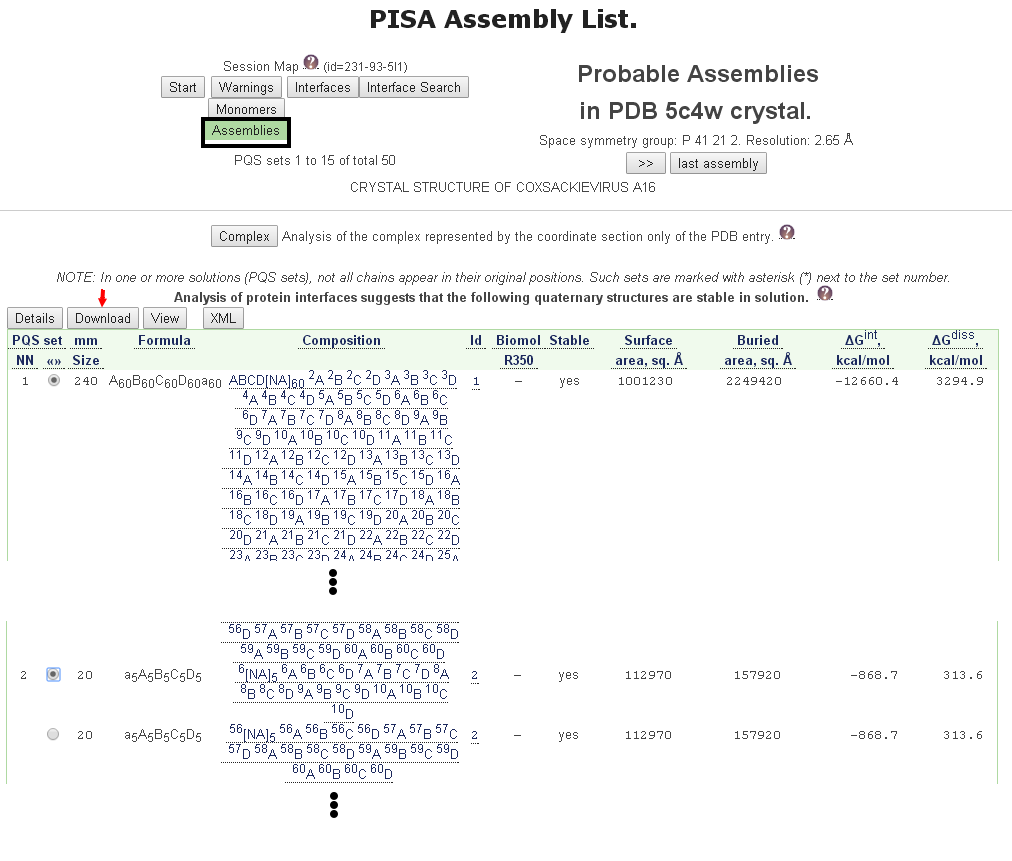
[圖十二]
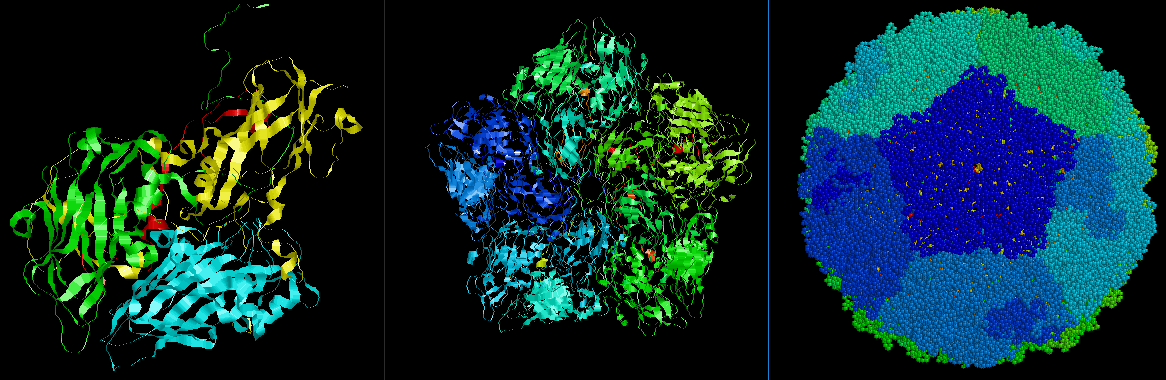
[圖十三]
雖然PISA是多年前發表的資料庫,儲存於PISA中的結構資料隨著PDBe (PDB in Europe)的增加而更新。因為它允許晶體封裝相互作用,所以在探討分子間的接合面時須謹慎使用,應選擇同一複合物的蛋白質鏈。另一方面,預設的結構視窗工具Jmol是基於Java Applet設計,但是瀏覽器Chrome已經不再支援此設計,所以在Windows平台僅能使用Internet Explorer瀏覽器,為安全起見,建議下載並使用其他結構視窗工具例如Rasmol、Swiss-PdbViewer或Pymol等等。
感謝生醫所黃明經老師實驗室孫慶姝小姐提供以上寶貴資料