本文經訊聯基因數位股份有限公司(原名:創源生技)同意轉載
全新RNA-seq 分析入口網站,搭配 Analysis Match Explorer 授權提供
我們推出全新的 RNA seq analysis portal ,讓資料分析更加簡單。 從 FASTQ
檔案到路徑分析,現在只需數小時!無論您使用 QIAseq 還是其他主流 RNA
文庫製備套件(如 Illumina 、 Thermo Fisher 、 NEB ),該入口皆能兼容並支援
20 種物種的分析,並運用已發表的主流算法進行處理。
透過這個網頁介面,您可以輕鬆將數據來自於 SRA 或是 BaseSpace 上傳至雲
端 (AWS),進行品質控制( QC),並將差異表達的結果直接推送至 IPA ,快速進行生物學解讀。 整個流程幾乎不需要人工干預,僅需 1-2 小時即可完成。
要使用此功能,您需擁有Analysis Match 和 Land Explorer 的授權。如果您具備這些授權,您將在 IPA 使用者頁面右上角找到 "Process RNA-seq data" 的按鈕,如圖 1 所示。
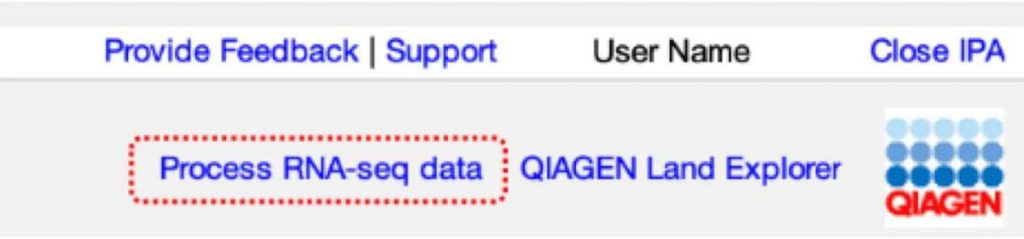
圖1:RNA-seq 分析介面連結 。 標題為 ”Process RNA-seq data” 的連結以紅
色虛線框顯示。 僅供持有 IPA 中 Analysis Match 和 Land Explorer 授權的
用戶使用。
RNA-seq portal 將引導您利用三個簡單的步驟分析與查看數據,如圖 2 所示
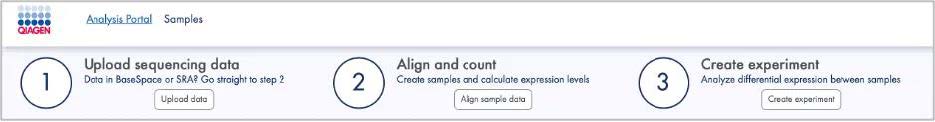
圖2:處理 RNA-seq 資料的三個簡單步驟。 1) 上傳, 2) 比對和計算表現亮,3) 實驗設計,利用實驗設計來比較出差異表現量 。
RNA-seq portal 分析流程
全新的RNA-seq portal 提供了一個直觀且高效的數據處理平台,從原始的
FASTQ 檔案開始,到最終差異基因表達分析,只需經過幾個簡單步驟即可完
成。以下是 RNA-seq 數據處理流程的詳細說明:
1.數據上傳與預處理
首先,您可以將原始的FASTQ 檔案從多種來源上傳至雲端( AWS )。無論您
的數據來自 QIAseq 、 Illumina 、 Thermo Fisher 或 NEB 等主流的 RNA 文
庫製備套件,該入口都能支持處理。此外,您還可以從 SRA Sequence
Read Archive )或 Illumina 的 BaseSpace 下載公開的研究數據,直接跳過
數據上傳步驟。
這一過程透過入口的網頁介面進行,無需複雜的命令操作,適合各種背景的研
究人員。系統會自動執行數據的品質控制 ,包括檢查數據完整性、讀取長度、
定序品質等。這些品質控制過程可確保後續分析的準確性。
2.對齊與計數
在數據上傳並通過初步品質控制後 RNA-seq portal 將使用已發表且驗證的對齊
算法(如 STAR 、 HISAT2 等)將您的讀數( reads )對齊至所選物種的參考
基因組。這個過程會自動化地進行,無需使用者干預。
完成比對 (alignment),系統會自動計算每個基因的讀數,生成 read counts 矩陣 並將基因表達數據進行歸一化處理,排除低表達背景訊號。這個高效的處理過程通常可在 1-2 小時內完成。
3.差異表達分析設置
完成對齊和計數後,您可以根據研究需求進行樣本分組(如基因敲除與對照
組 ),設定差異表達基因( DEGs )的分析條件。系統會使用 DESeq2 、 EdgeR
等算法,計算每個基因的 fold change 和 p 值,生成差異基因結果。分析完
成後,系統會自動生成火山圖、熱圖等視覺化結果,幫助您快速瀏覽基因差
異。 火山圖可以顯示顯著變化的基因,而熱圖則能幫助您觀察樣本間的基因表
達模式。此外, RNA-seq portal 還提供了 PCA (主成分分析) 圖,幫助您檢
查數據是否合理地群聚在各自的樣本組中(圖 4) 。這樣的視覺化工具可即時幫助
您進行 品質控制 與數據檢查,確保後續分析結果的可靠性。 完成這些步驟後,
您可以查看您的數據,如圖 3 所示
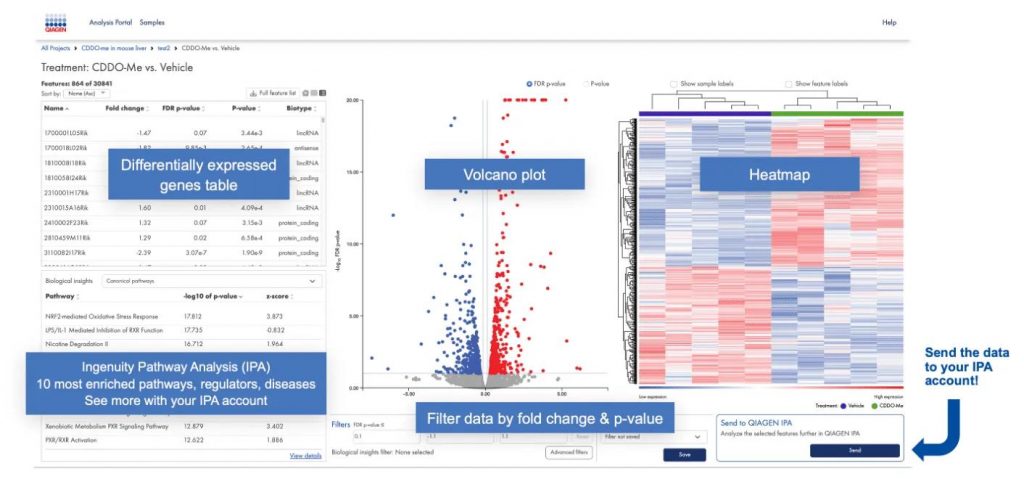
圖 3.RNA seq portal 「實驗視圖」的註釋視圖。將樣本根據實驗所設定
的分組條件,可以進行基因差異表現量的分析。並且將差異表現量基因已
熱圖與火山圖的方式進行可視化 。設定 閾值 ,您可以將資料傳送到您的
IPA 帳戶進行 路徑分析與生物解釋 。
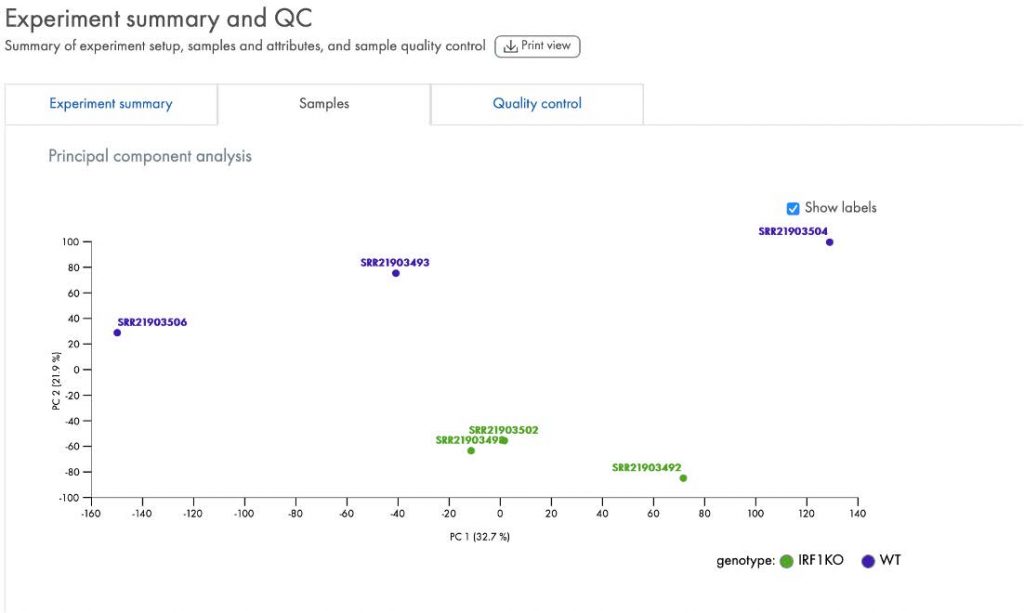
圖4 .PCA (主成分分析)。 每個樣本都標有屬於他們分組的關鍵屬性。理想情
況下,就像本例中的「基因型」一樣,樣本可以根據其基因型清晰地聚集。
PCA 結果可以顯示樣本是否確實存在由組別差異驅動的 RNA 表現量差異,例
如 IRF1 是否如本例所示被敲除(數據源自 GSE215771 )。
其他QC 工具
在RNA-seq Portal 也有 其他 QC 工具可依基因組區域(例如內含子、外顯子
或基因間區域)、生物型(圖 5 )或污染生物體的分類特徵(圖 6 )進行繪
製。
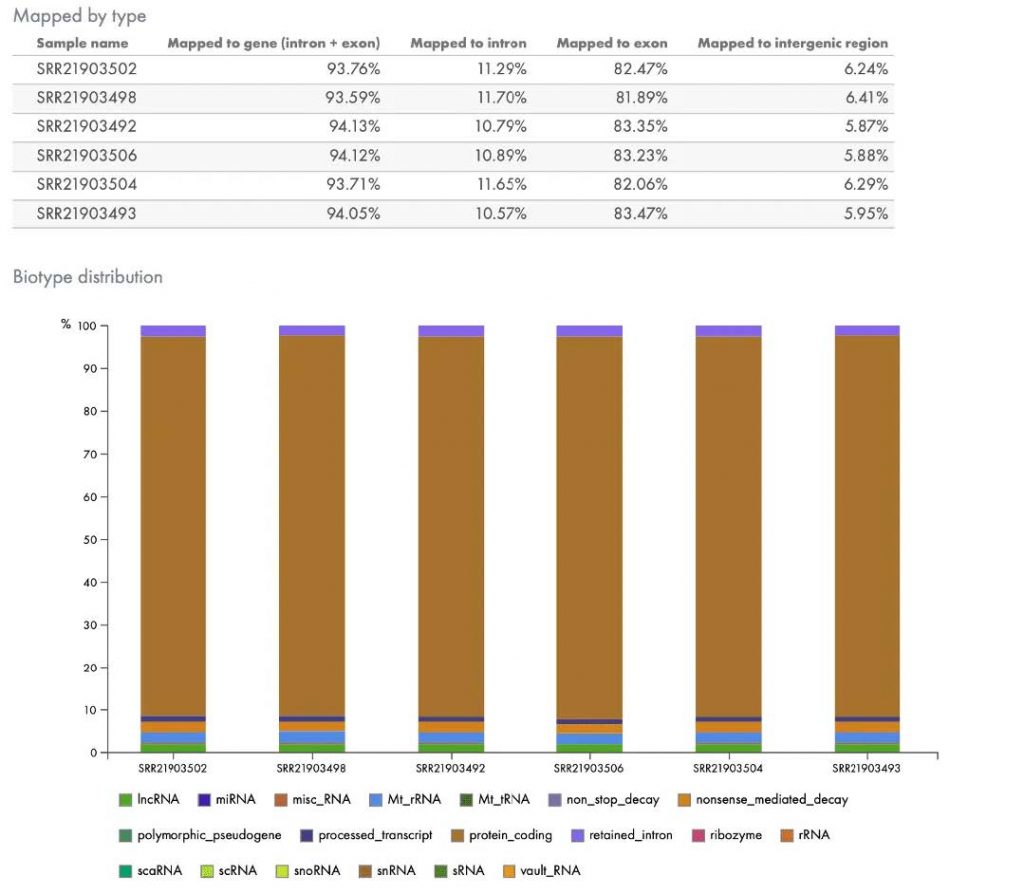
圖5 . 依基因組區域(上)或以生物型(下)劃分的 RNA 作圖 。 RNA-seq
portal 可以描述每一個樣本 讀取在基因組(廣義)中的位置(上圖),以及
RNA 類型(例如蛋白質編碼區或 lncRNA)的指標 下圖 。
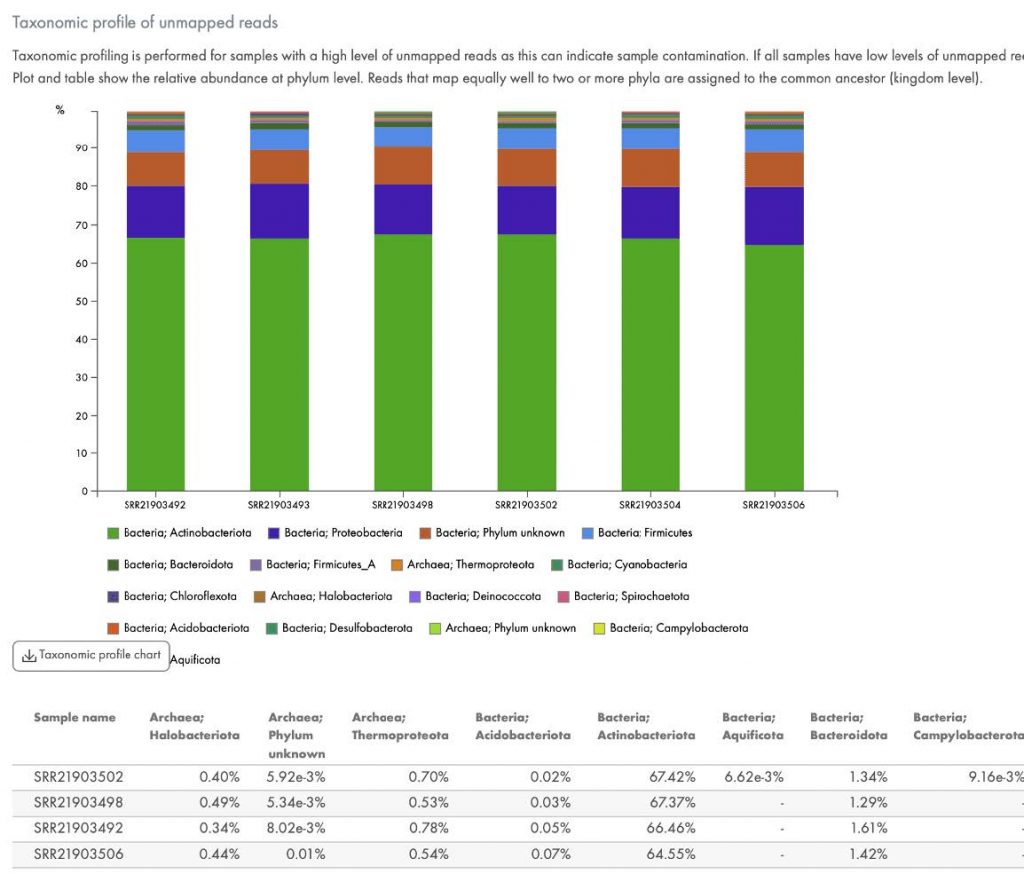
圖 6. 污染 RNA 的定量。如果您的樣本被細菌序列污染,將提供此圖表來定量細菌類型。
除了上述功能, RNA seq portal 還提供了多種數據共享選項。您可以與團隊成員分享數據集,生成文氏圖 Venn diagram) 來比較不同實驗的基因重疊情況,或將數據傳送至 IPA 中進行核心分析。這使得多個實驗間的比較與分析變得簡單 ,讓您輕鬆獲得關鍵的生物學模式。
修正的同源基因群集
IPA 已更新同源基因的定義,在此版本之前,IPA利用NCBI HomoloGene 定義同源基因,但HomoloGene已在2014年停止更新,現在轉而採用NCBI 真核基因組註釋流程(NCBI Eukaryotic Genome Annotation Pipeline)中的新方法。這項變更影響了大約 10% 的同源基因,新的同源基因定義優於舊的定義。對於已經更改的同源基因,舊的同源基因將視為”棄用”,過去的分析可能需要重新運行,以確保您能受益於更精確、最新的基因定義。可利用右鍵單擊分析並選擇“重新運行分析”。
氣泡圖改進
當您自訂典型路徑(canonical pathway)氣泡圖以路徑類別為氣泡著色時,Reactome Pathways 將與 Ingenuity Signaling 和 Metabolic 路徑分開著色,如下圖 7 所示。
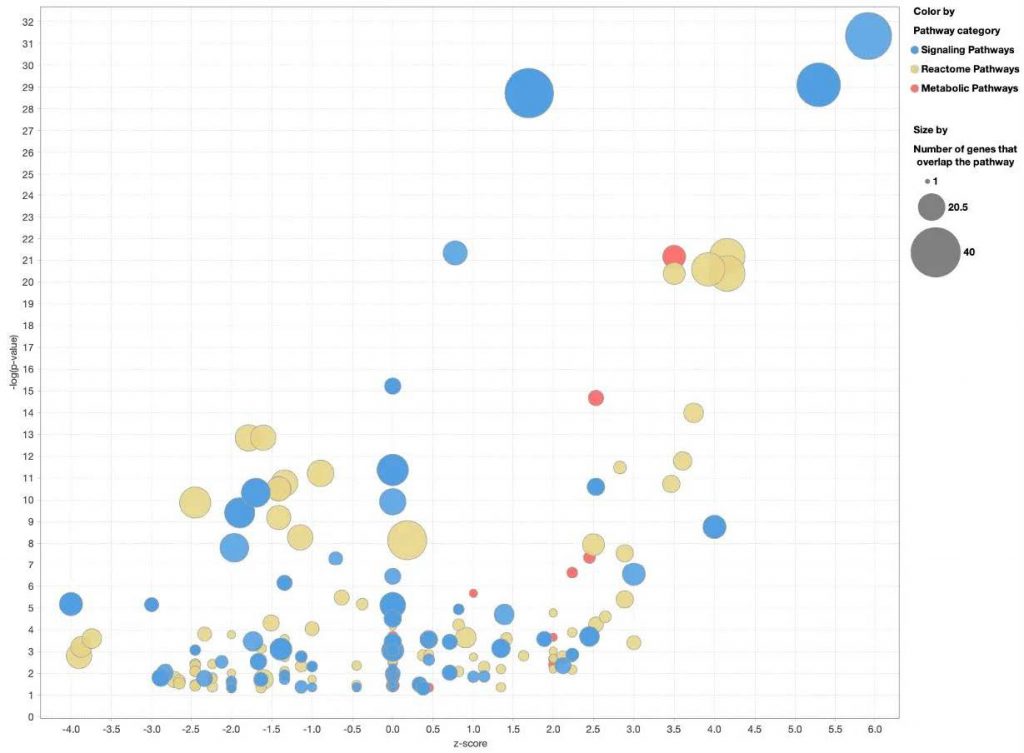
圖 7. 依路徑類別為 典型 路徑氣泡圖著色。現在, Reactome 路徑的顏色與
Ingenuity Signaling 和 Metabolic Pathways 不同 。
內容更新
11 個新的 Ingenuity 訊號路徑
• 親環蛋白訊號路徑(Cyclophilin Signaling Pathway)
• 糖化訊號路徑(Glycation Signaling Pathway)
• 乙型肝炎慢性肝臟發病機轉訊號路徑(Hepatitis B Chronic Liver Pathogenesis Signaling Pathway)
• 腸躁症訊號路徑(Irritable Bowel Syndrome Signaling Pathway)
• 肺離子平衡訊號路徑(Lung Ionic Balance Signaling Pathway)
• 粒線體分裂訊號路徑 (Mitochondrial Division Signaling Pathway)
• mRNA 3 端加工訊號路徑 (mRNA 3 Prime End Processing Signaling Pathway)
• NAP1L1 轉錄調控訊號路徑 (NAP1L1 Transcription Regulation Signaling Pathway)
• 核醣體品質控制訊號路徑 (Ribosomal Quality Control Signaling Pathway)
• Sheddase 訊號路徑 (Sheddase Signaling Pathway)
• TRIM21 細胞內抗體訊號路徑(TRIM21 Intracellular Antibody Signaling Pathway)
5 4 個 Re actome 路徑
GWAS 新發現
超過60,000項來自 NHGRI-EBI資料庫的人類全基因組關聯研究( GWAS)結果現
已整合到 IPA中。這些資料中,所有能夠與基因相關的關係(而非位於基因間區
域的變異)都已納入 IPA中的“開放存取的全基因組關聯結果資料庫”。同時,約
2,000項來自 2009年的發現因不符合當前的標準被移除。
如圖8所示,展示了一個使用這些 GWAS發現建立的小型網絡範例。僅包含 p值小於或等於 1e-05的關聯結果,且具體的 p值會在 IPA的未來版本中顯示。
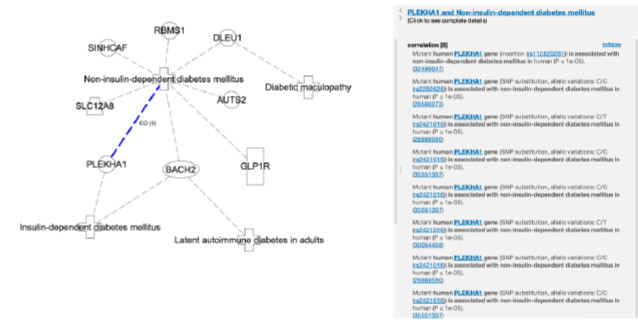
圖8.IPA 中新 GWAS 關聯關係的範例。
僅導入了p 值小於或等於 1e 05 的關聯結果。具體的 p 值資訊將在 IPA 的未來
版本中顯示。
更新機器學習疾病路徑
ML疾病途徑已透過最佳化、新內容和新直系 同源基因 定義重新計算。